Using the Python API#
In this interactive Jupyter notebook, you will learn how to build a simple random forest site-of-metabolism (SOM) classifier using a synthetic dataset. We also show how to use our utility for computing the FAME score, which is a similarity-based applicability domain score.
The resulting classifier shown here will be very similar to the classifier created in the CLI tutorial, but crucially you can use Python scripting and scikit-learn to modify the model to your liking, add new features, or use different model architectures.
Preparing the data#
from ast import literal_eval
from rdkit.Chem import Draw
from rdkit.Chem.rdmolfiles import SDMolSupplier
Let’s start by loading the synthetic metabolism dataset. Known SOM indices stored as SDF/MOL2 molecule properties are also extracted here.
molecules = [
mol for mol in SDMolSupplier("data/metatrans_autoannotated_cleaned/train.sdf")
]
soms = [literal_eval(mol.GetProp("soms")) for mol in molecules]
We can take a quick look at some of the molecules from the dataset, with known SOMs being highlighted in red.
Draw.MolsToGridImage(molecules[:16], highlightAtomLists=soms, molsPerRow=4)
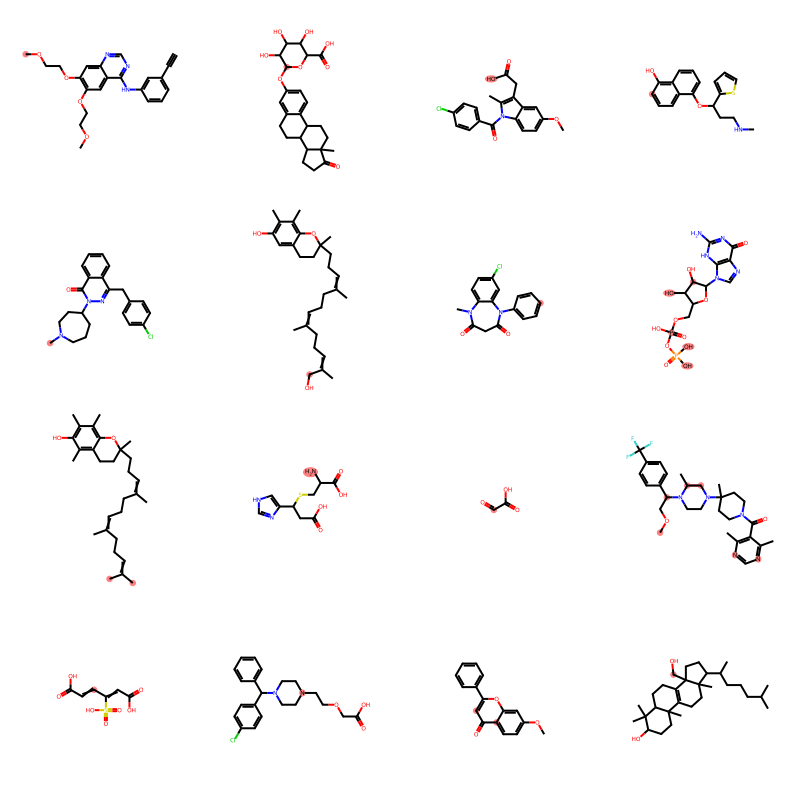
Now, we extract the individual atoms, along with their SOM label.
atoms_and_labels = [
(atom, atom.GetIdx() in soms)
for mol, soms in zip(molecules, soms)
for atom in mol.GetAtoms()
]
atoms, labels = zip(*atoms_and_labels)
RDKit helper function#
from rdkit.Chem.rdchem import Atom, Mol
from rdkit.Chem.rdmolfiles import MolToSmiles
In order to get our atoms into a format that FAME3R understands, we define a small helper function.
The function converts an RDKit atom into a SMILES string that uses atom mapping information to mark that exact atom.
def atom_to_marked_smiles(atom: Atom) -> str:
mol = Mol(atom.GetOwningMol())
mol.GetAtomWithIdx(atom.GetIdx()).SetAtomMapNum(1)
return MolToSmiles(mol)
Tip
If you are already using CDPKit in your code, you can directly pass Atom instances to fame3r.FAME3RVectorizer. This is great for efficiency, as no additional conversions need to be performed. However, you should be aware that Atom defines a __getitem__ method which interferes with normal NumPy array conversion, meaning you will have to pre-allocate an empty NumPy array and assign to it like so:
atom_array = np.empty((len(atoms), 1), dtype=object)
atom_array[:, 0] = atoms
Training the model#
from sklearn.ensemble import RandomForestClassifier
from sklearn.pipeline import make_pipeline
from fame3r import FAME3RVectorizer
We can use the scikit-learn sklearn.pipeline.Pipeline utility to create a simple random forest model and train it on our synthetic dataset.
The fame3r.FAME3RVectorizer is used to generate the FAME3R descriptors the random forest model is trained on.
The hyperparameters chosen are the same as the defaults used in the fame3r CLI tool.
Tip
The fame3r.FAME3RVectorizer is configurable using various keyword arguments. Look into our API documentation to customize it to your needs.
model = make_pipeline(
FAME3RVectorizer(input="smiles", radius=5),
RandomForestClassifier(
n_estimators=250,
max_depth=None,
min_samples_split=2,
min_samples_leaf=1,
max_features="sqrt",
class_weight="balanced_subsample",
n_jobs=-1,
),
)
model.fit([[atom_to_marked_smiles(atom)] for atom in atoms], labels)
Pipeline(steps=[('fame3rvectorizer', FAME3RVectorizer()),
('randomforestclassifier',
RandomForestClassifier(class_weight='balanced_subsample',
n_estimators=250, n_jobs=-1))])In a Jupyter environment, please rerun this cell to show the HTML representation or trust the notebook. On GitHub, the HTML representation is unable to render, please try loading this page with nbviewer.org.
Parameters
| steps | [('fame3rvectorizer', ...), ('randomforestclassifier', ...)] | |
| transform_input | None | |
| memory | None | |
| verbose | False |
Parameters
| radius | 5 | |
| input | 'smiles' | |
| output | ['fingerprint', 'physicochemical', ...] |
Parameters
| n_estimators | 250 | |
| criterion | 'gini' | |
| max_depth | None | |
| min_samples_split | 2 | |
| min_samples_leaf | 1 | |
| min_weight_fraction_leaf | 0.0 | |
| max_features | 'sqrt' | |
| max_leaf_nodes | None | |
| min_impurity_decrease | 0.0 | |
| bootstrap | True | |
| oob_score | False | |
| n_jobs | -1 | |
| random_state | None | |
| verbose | 0 | |
| warm_start | False | |
| class_weight | 'balanced_subsample' | |
| ccp_alpha | 0.0 | |
| max_samples | None | |
| monotonic_cst | None |
Making predictions#
from rdkit.Chem import Draw
from rdkit.Chem.rdmolfiles import MolFromSmiles
We can now use the trained model to make predictions about individiual atoms.
For example, consider two atoms in morphine. First, the oxygen of the hydroxyl group attached to C3, and second the quarternary carbon C12.
morphine = MolFromSmiles("CN1CC[C@]23C4=C5C=CC(O)=C4O[C@H]2[C@@H](O)C=C[C@H]3[C@H]1C5")
Draw.MolToImage(morphine, highlightAtoms=[10, 4])
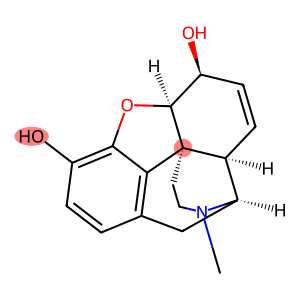
The model predicts that the considered hydroxyl group would be much more reactive than the quarternary carbon. This matches basic chemical intuition, as well as the known metabolism pathway of opiates.
model.predict_proba(
[
[atom_to_marked_smiles(morphine.GetAtomWithIdx(10))],
[atom_to_marked_smiles(morphine.GetAtomWithIdx(4))],
],
)[:, 1]
array([0.936, 0. ])
Using the FAME score#
from rdkit.Chem import Draw
from rdkit.Chem.rdmolfiles import MolFromSmiles
from sklearn.pipeline import make_pipeline
from fame3r import FAME3RScoreEstimator, FAME3RVectorizer
The FAME score is a similarity-based applicability domain score. It ranges from 0 to 1 with higher values indicating the considered atom environment is more well-represented in the training data.
We train a model for calculating that score based on atoms in the dataset in much the same way as our classification model. Crucially, we only use fingerprint descriptors here, as the FAME score is well-defined only for binary features.
applicability_domain = make_pipeline(
FAME3RVectorizer(input="smiles", radius=5, output=["fingerprint"]),
FAME3RScoreEstimator(n_neighbors=3),
)
applicability_domain.fit([[atom_to_marked_smiles(atom)] for atom in atoms], labels)
Pipeline(steps=[('fame3rvectorizer', FAME3RVectorizer(output=['fingerprint'])),
('fame3rscoreestimator', FAME3RScoreEstimator())])In a Jupyter environment, please rerun this cell to show the HTML representation or trust the notebook. On GitHub, the HTML representation is unable to render, please try loading this page with nbviewer.org.
Parameters
| steps | [('fame3rvectorizer', ...), ('fame3rscoreestimator', ...)] | |
| transform_input | None | |
| memory | None | |
| verbose | False |
Parameters
| radius | 5 | |
| input | 'smiles' | |
| output | ['fingerprint'] |
Parameters
| n_neighbors | 3 |
As an example, we can consider omeprazole. The molecule contains a sulfoxide group, a relatively rare structural feature, as well as an methoxy group, which is very commonly found in our dataset. In addition, the sulfoxide group is in close proximity to a
omeprazole = MolFromSmiles("CC1=CN=C(C(=C1OC)C)CS(=O)C2=NC3=C(N2)C=C(C=C3)OC")
Draw.MolToImage(omeprazole, highlightAtoms=[11, 23])
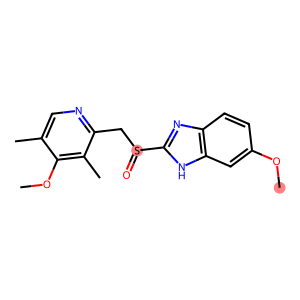
As expected, the sulfur atom of the rarer sulfoxide group receives a much lower FAME score than the methyl carbon of the more common ether group.
applicability_domain.predict(
[
[atom_to_marked_smiles(omeprazole.GetAtomWithIdx(11))],
[atom_to_marked_smiles(omeprazole.GetAtomWithIdx(23))],
],
)
array([0.61913043, 0.92592593])